Autosomal Dominant spinocerebellar ataxia - case presentation .
Case Description
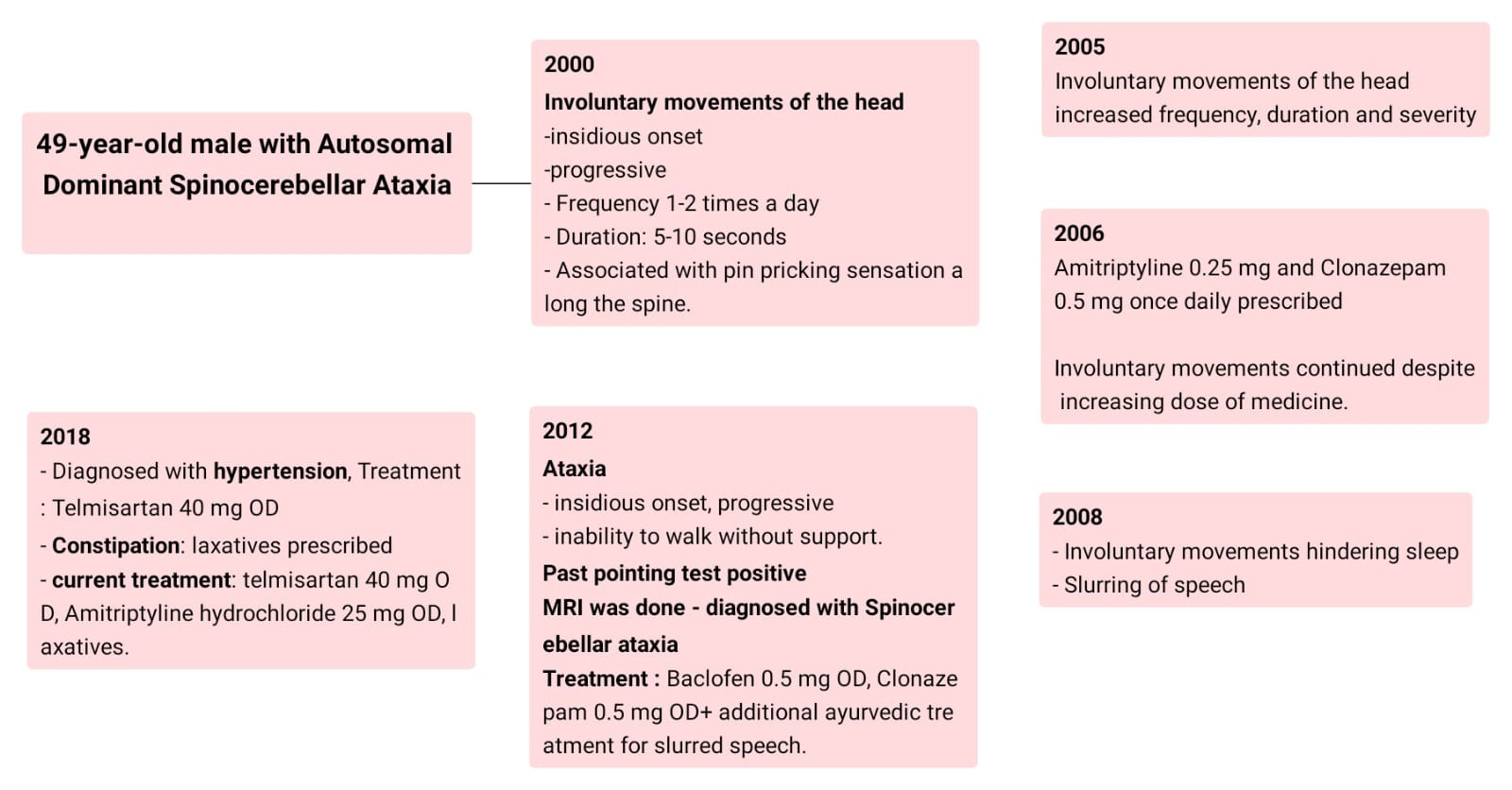
A 49-year-old male, with a history of hypertension, a businessman by occupation, presented with involuntary movements of the head.
The patient was apparently asymptomatic till the year 2000, while
watching television, he felt the back of the neck and head become stiff, and soon after,
he experienced involuntary movements of his head, moving from
side to side (The patient described It as if he was saying no with the movement
of his head alone). They were insidious in onset and progressive in nature. Initially, the involuntary movements were visible only on careful observation. It lasted 5 to 10 seconds and happened 1-2 times a day, a few times a week. The involuntary
movements of the head were associated with a pin-pricking sensation
along his spine.
By the year 2005, the involuntary movements of the head
increased in frequency, duration and severity such that the patient was unable to
complete the task at hand. At this point, the patient visited a local physician
but the medications did not provide relief.
In 2006, the patient visited a higher centre, where the
doctor prescribed Amitriptyline 0.25 mg and Clonazepam 0.5 mg once daily. These
medications provided some relief for 3 months. After this the involuntary
movements continued even after increasing the doses of medicines. The patient also tried homoeopathic medication,
but it did not provide any relief.
In 2008, the patient started experiencing reduced sleep,
whenever he tried to rest, in the the supine position, the involuntary movements of the
head reappeared, thus he had to lie down on his sides. The patient also started
experiencing slurring of speech.
By the year 2012, the patient developed ataxia which
was insidious in onset and progressive in nature. In a year, the patient was unable
to walk without support. past pointing test was positive. He visited a
doctor where an MRI was done and the patient was diagnosed with Autosomal
Dominant Spinocerebellar degeneration. The patient was treated with Baclofen 5mg
and clonazepam 0.5 mg. The patient experienced some relief from symptoms for a
few months. He also started ayurvedic treatment, which was found beneficial, for
slurred speech -which included chewing the roots of the plant piper longum. The
patient stopped taking medication as it was not providing any relief.
In 2018 the patient was diagnosed with hypertension and was
started on telmisartan 40 mg Once Daily. The patient also complained of
constipation and consumes laxatives occasionally. The patient was started on
Amitriptyline Hydrochloride 25 mg OD for symptomatic management.
There is no history of muscle weakness, abnormal eye movements,
dysarthria, loss of sensations, or visual or hearing impairment.
There are no features of parkinsonism, epileptic seizures, or myoclonus.
The current treatment includes Amitriptyline Hydrochloride
25 mg OD, telmisartan 40 mg OD and physiotherapy.
Family History

![]()
Father: No known medical issue. Died
in 2003.
Mother: diagnosed with Autosomal
dominant Spinocerebellar degeneration at the age of 34 and passed away at the
age of 35
Elder sister: Died in 2003 due to
autosomal dominant spinocerebellar ataxia at the age of 41
Elder brother: diagnosed with
Autosomal dominant Spinocerebellar degeneration in 2003, died: in 2013 at the age of
42
Younger sister: Died: in 2021 due to
autosomal dominant spinocerebellar ataxia at the age of 56
Wife: No medical issues
Daughter: No symptoms of disease till
now.
Case discussion:
This case depicts the clinical progression of the disease.
Autosomal
dominant spinocerebellar ataxias are progressive in nature characterised by slow
degeneration of the cerebellum accompanied by degeneration of other parts of
the central nervous system including the brainstem. [1]
A
patient is known to have a genetic form of ataxia when there is
-
History of
insidious onset
-
Slow
progression
-
Bilaterally
symmetrical findings on examination
-
Positive family
history of ataxia in the patient’s parents. 1
In our patient,
the patient’s mother was diagnosed with Autosomal Dominant Spinocerebellar Ataxia.
and the history of ataxia is consistent with the above findings.
The global prevalence of spinocerebellar ataxia is noted as
3 in 100,000 [3]
Though prevalence studies in all countries note idiopathic
cases of Spinocerebellar degeneration. Median Proportions of Patients with
Spinocerebellar degenerations with unknown aetiologies include 38.5 per cent in
India. While SCA2 is the most common subtype prevalent in India [2]
A study to find the severity and survival in
Inherited spinocerebellar ataxias found that survival was 68 years in 223
patients with polyglutamine expansions, versus 80 years in 23 patients
with other mutations. Disability was also more severe in patients with
polyglutamine expansions.[4]
Clinical Features
All patients with this disease present with cerebellar ataxia,
but the additional symptoms are variable. [5]
For Instance, according to the gene involved – [1]
|
Involved gene |
Clinical
features |
|
SCA1 |
signs of widespread cerebellar and brainstem
dysfunction with relatively little supratentorial involvement. |
|
SCA2 |
ataxia, dysarthria, slow saccades, and peripheral
neuropathy |
|
SCA3 |
progressive ataxia, lid retraction,
infrequent blinking, ophthalmoparesis, impaired speech and swallowing |
|
SCA5 |
a relatively pure form of slowly progressive dominant cerebellar
ataxia |
|
SCA6 |
pure” cerebellar ataxia accompanied by
dysarthria and gaze-evoked nystagmus with onset at 20 years. |
|
SCA7 |
the universal presence of retinal degeneration with ataxia |
|
SCA8 |
Prominent gait and limb ataxia, abnormalities of swallowing,
speech and eye movements |
|
SCA10 |
prominent cerebellar symptoms and
seizures. |
|
SCA11 |
pure” cerebellar syndrome with mild pyramidal signs slowly progressive form of gait and limb ataxia |
SCA12
|
more common in India Age at onset ranges between 8 to 60 years first symptom typically being an action tremor of the arms.
The tremor is eventually accompanied by head tremor, ataxia, and sometimes
bradykinesia and sensory neuropathy. |
SCA13
|
widely varying ages of onset Common features are ataxia, dysarthria, nystagmus, and occasionally
hyperreflexia. |
SCA14
|
slowly progressive ataxia with dysarthria in early adulthood In late-onset cases, SCA14 can manifest as a relatively pure
cerebellar ataxia |
SCA15/16
|
slowly progressive, pure cerebellar ataxia Dysarthria, horizontal gaze-evoked nystagmus, and impaired
smooth movement of the eyes are present in some patients. Approximatelyone-thirdd of patients have a head tremor. The disease is caused by small genomic
deletions encompassing the IPTR |
SCA17
|
young-adulthood or mid-adulthood progressive gait and limb ataxia dementia, psychiatric symptoms, and varying
extrapyramidal features, including parkinsonism, tremor, dystonia, and
sometimes chorea, seizures |
SCA20
|
initial symptom is
dysarthria rather than gait ataxia, accompanied by palatal tremor,
hypermetric saccades, and dentate calcification in the cerebellum |
SCA27
|
early-onset ataxia manifest first
with handtremorsr in childhood followed by progressive ataxia, cognitive
difficulties, and psychiatric problems in the second and third decades of
life. |
According to the following findings, the clinical features
in our patient are most consistent with SCA12.
MRI and genetic testing in addition to a through family
history are usually done in order to diagnose a patient with Autosomal Dominant
Spinocerebellar ataxia. On the MRI for diagnosis there should be presence of
cerebellar atrophy and evidence of structural or vascular damage and or other
lesions or associated neurodegeneration. [5] Our patient underwent MRI in the
year 2012, and was diagnosed with Inherited Spinocerebellar ataxia, but due to
lack of documentation, the patient lost the reports.
Genetic testing can be done in 5 distinct scenarios – diagnostic testing,
predictive testing, prenatal testing carrier testing, and risk factor assessment. In reality, however, only
diagnostic and predictive testing concern the practicing physicians. [1]
On the basis of genetic testing various SCA genes are identified, and
clinical features are noted as depicted in the table above. However, in our
patient the genetic testing was not done.
There is presence of very strong family history in our patient
consistent with autosomal dominant spinocerebellar ataxia.
There is no effective treatment for
genetic and idiopathic ataxia, hence includes symptomatic treatment an exercise
therapy to maintain patient function. [5]
This was incorporated in the management
of our patient as well.
Learning Outcomes
The following consultation occurred as telecommunication, A
PAJR group was created. This group aimed at understanding the patient’s
problems and providing the best possible solution in terms of pharmacotherapy
and exercise therapy. In addition to this the patient also found a way to describe
his day-to-day activities which added to the accountability of the patient to
maintain a healthy diet, compliance with exercise and pharmacotherapy was
checked on a daily basis.
SWOT analysis of this approach is as follows.
Strength:
-
The patient found a means of communication, to
express his difficulties on a day-to-day basis, these were immediately managed.
-
The group provided a way to determine the family
history which was very essential in the diagnosis.
-
The PaJR group acted as a means of compliance in
terms of exercise and pharmacotherapy.
Weakness
-
There was lack of face-to-face communication and
a lack of human touch acted as a major drawback especially in a disease wherein,
the prognosis is not good.
-
Language barrier: As the patient speaks a
different language than most doctors on the PaJR group, it hindered in
immediate management.
-
Lack of investigations and lost reports were a
major drawback.
Opportunities:
-
This approach allowed a detailed history and an
understanding of how the disease affected the individual in terms of day-to-day
activities
-
It acted as a platform for medical students to
directly see how a patient would present with this disease
-
It also acted as a platform for doctors to
discuss treatment and research modalities.
Threats:
-
Inability for members of the group to understand
the patient’s problems due to a language barrier.
References
1.
Paulson,
H. L. (2009). The spinocerebellar ataxias. Journal of
Neuro-Ophthalmology: The Official Journal of the North American
Neuro-Ophthalmology Society, 29(3), 227–237. https://doi.org/10.1097/WNO0b013e3181b416de
2.
van
Prooije, T., Ibrahim, N. M., Azmin, S., & van de Warrenburg, B. (2021).
Spinocerebellar ataxias in Asia: Prevalence, phenotypes and management. Parkinsonism
& Related Disorders, 92, 112–118.
https://doi.org/10.1016/j.parkreldis.2021.10.023
3.
Luis RuanoClaudia MeloM. Carolina SilvaPaula Coutinho; The Global Epidemiology of
Hereditary Ataxia and Spastic Paraplegia: A Systematic Review of Prevalence
Studies. Neuroepidemiology 1
April 2014; 42 (3): 174–183. https://doi.org/10.1159/000358801
4.
Monin, L., Marelli, C., Cazeneuve, C., Charles,
P., Tallaksen, C., Forlani, S., Stevanin, G., Brice, A., & Durr, A. (2015).
Survival and severity in dominant cerebellar ataxias. Annals of Clinical and
Translational Neurology, 2(2), 202-207. https://doi.org/10.1002/acn3.156
5.
Shakkottai, V. G., & Fogel, B. L. (2013).
Autosomal Dominant Spinocerebellar Ataxia. Neurologic clinics, 31(4).
https://doi.org/10.1016/j.ncl.2013.04.006
6. Link to first case report - https://ssahamedicalcases.blogspot.com/2023/02/patient-history-pt-is-49-yrs-old-male.html?m=1
 |
|||
 |
|||


Comments
Post a Comment